15% Early Bird discount until 31 Oct 2025
IVDR Medical Device Conformity Assessment Training
Practice-oriented, 10-month training
Starting Date: 30 January 2026

Az IVDR előírja, hogy a gyártóknak az eszköz teljes életciklusa alatt teljesítőképességi értékelést kell végezniük annak érdekében, hogy az eszköz biztonságossága és teljesítőképessége megfeleljen a rendeletben foglalt követelményeknek, és ezáltal a betegek és felhasználók számára egyaránt biztosított legyen a rendeltetési célnak történő megfelelés és az előny–kockázat elfogadható aránya.
A gyártónak minőségirányítási rendszert (MIR) kell bevezetnie, valamint működtetnie és rendelkeznie kell forgalomba hozatal utáni felügyeleti rendszerrel (Post Market Surveillance System, továbbiakban: PMS), hogy megfeleljen a rendelet követelményeinek.
A PMS-nek az adott eszköz kockázati osztályával arányosnak kell lennie. A PMS mellett a gyártóknak kockázatkezelési rendszert is létre kell hozniuk, amely alkalmas arra, hogy az eszközzel kapcsolatos váratlan eseményeket dokumentálja, jelentse és a szükséges intézkedéseket elvégezze.
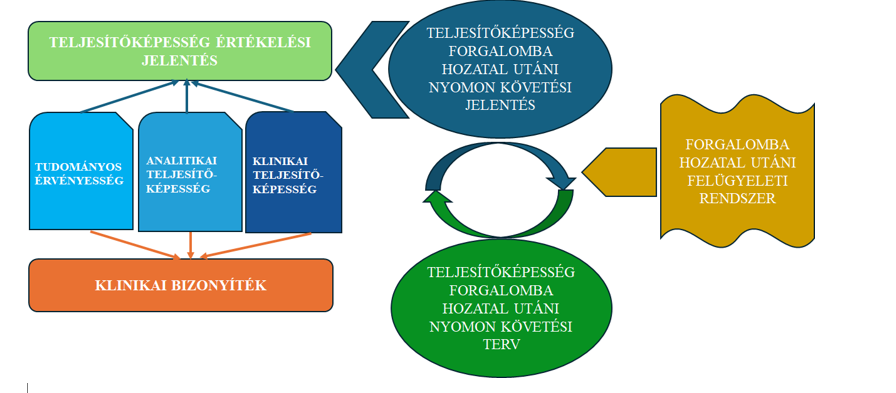
1. ábra: A PMS és a teljesítőképesség-értékelés kapcsolata
A gyártónak a forgalomba hozatal utáni felügyeleti rendszerében többek között az alábbi feladatok elvégzése céljából proaktív módon kell adatokat gyűjtenie (részletesen a VII. fejezetben):
Az IVDR XIII. mellékletének B. része írja elő a teljesítőképesség forgalomba hozatal utáni nyomon követésével (Post Market Performance Follow Up, továbbiakban: PMPF) kapcsolatos követelményeket.
A PMPF (a teljesítőképesség forgalomba hozatal utáni nyomon követése, angolul: Post-market performance follow-up) célja, hogy a biztonságosság és a teljesítőképesség folyamatos ellenőrzése mellett a felmerülő kockázatok felderítése is megtörténjen.
Az IVDR új követelményként írja elő a PMPF-tevékenységet a gyártók számára (hasonlóan az MDR-ben szereplő forgalomba hozatal utáni klinikai nyomon követéshez, a PMCF-hez).
A PMPF-tervben meg kell adni a nyomon követés tervezett időpontjait, vagy bizonyos okok miatt soron kívüli időpontban kell elvégezni a PMPF-tevékenységet, és erről jelentést kell készítenie.
A PMPF elvégzésének gyakoriságát a gyártónak kell meghatároznia a kockázati osztály figyelembevételével, és ezt indokolnia is szükséges.
Az IVDR előírja, hogy a C. és D. osztályba tartozó eszközök esetében a PMPF-jelentést évente kell frissíteni.
A PMPF-tervben tételesen fel kell sorolni a releváns harmonizált szabványokra való hivatkozást, például, ha sterilitásra, csomagolásra vagy metrológiai nyomon követésre vonatkozó előírások alkalmazandók az eszközre.
A PMPF-tervben meg kell határozni az eszköz piacra kerülését követően a további adatok gyűjtésének célját, például, hogy a tudomány fejlődése, a megváltozott klinikai gyakorlat vagy új gyógyszerek bevezetése miatt a PMPF során megvizsgálja, van-e az új adatoknak és információknak befolyásuk az eszköz teljesítőképességére és biztonságosságára.
A PMPF-tervben le kell írni a konkrét módszereket és eljárásokat, ezek megfelelőségének indoklását, valamint a célkitűzést és a gyakoriságot/ütemezést. Ilyen lehet pl. genetikai vizsgálatok esetén olyan új mutációk megjelenése a szakirodalomban, vagy olyan gyógyszerek által kiváltott interferencia lehetősége merül fel az analittal, amely megváltoztathatja az eszköz teljesítőképességét. Ezek az új információk indokolhatják az eszköz analitikai vagy klinikai teljesítőképességének újraértékelését is, ezáltal biztosítva az eszköz folyamatos biztonságosságának és teljesítőképességének értékelését a teljes életciklus alatt.
A PMPF-terv tartalmi követelményei:
Példák a PMPF-tervezéshez:
Általános módszerek és eljárások | Speciális módszerek és eljárások | A módszer és eljárás megfelelőségének indoklása | Célkitűzések | Gyakoriság / ütemezés |
Szakirodalom–kutatás | Eljárási utasítás az irodalomkutatásra | Új tudományos információk gyűjtése az adott biomarkerre vonatkozóan | Az új variáns vizsgálata | Rendszeres szakirodalmi áttekintés |
Felhasználók visszajelzése | A kapott adatok és az eszköz használata során bekövetkezett esemény kapcsolatának vizsgálata | Kapcsolódik-e a panasz az eszköz teljesítőképességéhez? | A szenzitivitás és specificitás javítása | A bekövetkezett eseménytől függően |
A PMPF-tervben meg kell határozni, hogy a proaktív módon történő adatgyűjtés és értékelés milyen célokat szolgál az eszköz biztonságosságára és teljesítőképességére vonatkozóan:
A PMS során gyűjtött adatokat és információkat, valamint a megelőző vagy korrekciós intézkedésekből levont következtetéseket, pl. kockázatkezelési tevékenység módosítását, a műszaki dokumentációban folyamatosan frissíteni kell.
Előfordulhat bizonyos esetekben, amikor a PMPF nem tekinthető követelménynek az IVDR alapján, például:
A PMPF eredményeit a PMPF értékelő jelentésben kell összefoglalni, melynek következtetéseket is kell tartalmaznia az alábbi dokumentációkra vonatkozóan:
A PMPF-tevékenység hasznos lehet a gyártó számára abban az esetben is, ha az eszköz rendeltetését szeretné kiterjeszteni, mivel előzőekben korlátozott rendeltetési célt határozott meg. A PMPF alkalmas lehet az eszköz továbbfejlesztésére és további bizonyítékok megszerzésére abból a célból, hogy a szélesebb körű használat lehetőségét igazolja, pl.:
Az IVD-eszköz teljesítőképességének forgalomba hozatal utáni nyomon követése egy folyamatos tevékenység, amelynek részei:
Az értékelés során megállapított következtetések és szükség szerinti megelőző vagy korrekciós intézkedések elvégzése biztosítja, hogy az eszköz teljes életciklusa alatt a rendeltetési célnak, a biztonságossági és teljesítőképességi követelményeknek megfeleljen.
Szerző: Tauberné dr. Jakab Katalin (Klinikai szakértő, NoBoVersum)
Részlet a NoBoVersum Zrt. IVDR orvostechnikai megfelelőségértékelő képzése tananyagából.
Practice-oriented, 10-month training
Starting Date: 30 January 2026