15% Early Bird discount until 31 Oct 2025
IVDR Medical Device Conformity Assessment Training
Practice-oriented, 10-month training
Starting Date: 30 January 2026

The IVDR requires manufacturers to conduct performance evaluations throughout the device lifecycle to ensure that the safety and performance of the device meets the requirements of the Regulation, thereby ensuring that it meets its intended purpose and provides an acceptable risk-benefit balance for both patients and users.
The manufacturer must implement a quality management system (MIR) and operate and maintain a post-market surveillance system (PMS) to meet the requirements of the Regulation.
The PMS must be proportionate to the risk class of the device. In addition to the PMS, manufacturers must also establish a risk control system capable of documenting and reporting any incidents related to the device and taking the necessary actions.
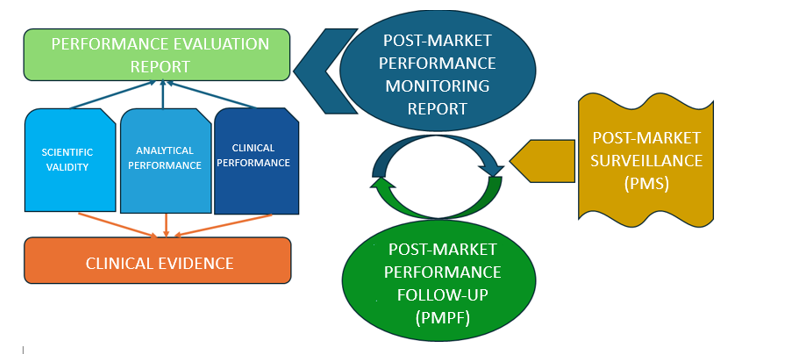
Figure 1: Relationship between PMS and performance evaluation
The manufacturer shall proactively collect data in his post-market surveillance system to perform, inter alia, the following tasks (detailed in Chapter VII):
Part B of Annex XIII to the IVDR sets out the requirements for Post-Market Performance Follow-Up (PMPF).
The PMPF (Post-Market Performance Follow-Up) aims to ensure that, in addition to the continuous monitoring of safety and performance, emerging risks are identified.
The IVDR introduces a new requirement for manufacturers to carry out PMPF activities [similar to the Post-Market Clinical Follow-up (PMCF) in the MDR].
The PMPF plan must specify the planned dates of follow-up or, for certain reasons, must be carried out at an out-of-sequence date and report on this.
The frequency with which the PMPF is to be carried out should be determined by the manufacturer, taking into account the risk class, and should be justified.
The IVDR requires that the PMPF report for Class C and D devices be updated annually.
The PMPF plan must list the reference to relevant harmonised standards, for example, if sterility, packaging or metrological traceability requirements apply to the device.
The PMPF plan shall specify the purpose of collecting additional data after the device has been placed on the market, for example, to assess whether new data and information, due to advances in science, changes in clinical practice or the introduction of new medicinal products, will have an impact on the performance and safety of the device during the PMPF.
The PMPF plan shall describe the specific methods and procedures, the justification for their conformity, the objective and the frequency/schedule. This may include, for example, the emergence of new mutations in the literature in the case of genetic testing, or the possibility of drug-induced interference with the analyte that could alter the performance of the device. This new information may also justify a re-evaluation of the analytical or clinical performance of the device, thus ensuring a continuous assessment of the safety and performance of the device throughout its lifecycle.
Content requirements of the PMPF plan:
Examples for PMPF design:
|
General methods and procedures |
Specific methods and procedures |
Justification of the conformity of the method and procedure |
Objectives |
Frequency /schedule |
|
Literature search |
Procedural instructions for literature search |
Collection of new scientific information on the biomarker in question |
Investigation of the new variant |
Regular literature review |
|
Feedback from users |
Examination of the relationship between the data obtained and the event occurring during the use of the tool |
Is the complaint related to the performance of the device? |
Repairing sensitivity and specificity |
Depending on the event that occurred |
The PMPF plan should specify the objectives of proactively collecting and evaluating data on the safety and performance of the device:
The data and information collected during the PMS and the conclusions drawn from preventive or corrective actions, e.g. modification of risk control activities, should be continuously updated in the technical documentation.
There may be certain cases where a PMPF is not considered a requirement under the IVDR, for example:
The results of the PMPF shall be summarised in the PMPF assessment report, which shall also include conclusions on the following documentation:
The PMPF activity may also be useful for the manufacturer in the case of an extension of the intended purpose of the device, as a limited intended purpose has been previously decided. The PMPF can be used to further develop the device and to obtain additional evidence to demonstrate the possibility of a wider use, e.g:
Post-Market Performance Follow-Up (PMPF) is an ongoing process of which:
The conclusions drawn from the assessment and the implementation of preventive or corrective actions, as appropriate, will ensure that the device meets its intended purpose, safety and performance requirements throughout its lifecycle.
Author: Katalin Tauberné Jakab (Clinical Expert, QTICS)
Extract from the NoBoVersum Zrt. IVDR Medical Conformity Assessment Training
Practice-oriented, 10-month training
Starting Date: 30 January 2026